7 Python interoperability
7.1 Introduction
Methods discussed in this book are usually available as either R or Python packages. Ideally, users should be able to leverage the full range of available tools for their analyses. Methods should be selected based on scientific merit (ideally demonstrated by neutral benchmarks), and independent of having been implemented in a given programming language (R or Python) or framework (e.g. Bioconductor or Seurat).
For single-cell and spatial omics data analysis, being able to leverage different ecosystems is especially powerful. On the one hand, Python offers superb infrastructure for image analysis and machine learning-based approaches. On the other hand, the R programming language has been historically dedicated to statistical computing; as a result, many modern R methods for spatial omics data build on a solid foundation of tools for spatial statistics and statistical modeling in general.
Different data structures, although standardized within a given framework, make switching between languages and tools somewhat cumbersome. In the realm of single-cell and spatial omics, all Bioconductor tools are built around SummarizedExperiment-derived classes, while Seurat (Hao et al. 2023), Giotto (Chen et al. 2025), and VoltRon each rely on their own object definitions. In Python, Scanpy (Wolf, Angerer, and Theis 2018) and Squidpy (Palla et al. 2022) use AnnData. Attempts to alleviate the problem are being made – e.g. zellkonverter, anndataR (Deconinck et al. 2025), and functions from Seurat allow for conversion between Python’s AnnData and R/Bioconductor’s SingleCellExperiment or SpatialExperiment.
On a higher level, tools that enable interoperability between programming languages have become available. For example, reticulate provides an R interface to Python, including support to translate between objects from both languages; basilisk facilitates Python environment management within the Bioconductor ecosystem, and can also be interfaced with reticulate; and Quarto can generate dynamic reports from code in different languages.
In this chapter, we demonstrate examples showing how to set up Python environments and interact with anndata objects in R, using either a virtual or Conda environment, together with reticulate or basilisk and anndataR.
7.2 Dependencies
7.3 Python environment and reticulate
We here set up a virtual environment to interact with Python anndata objects in R. In order to build this book programmatically, we install Python and pip dependencies from scratch using reticulate.
However, if you are following this example on a laptop or another system, it may be easier to specify an existing Python installation to use instead. This will avoid cluttering your system with additional Python installations; see the below section to, e.g., activate an existing Conda environment.
7.3.1 Set up virtual environment
First, we create a virtual environment, installing Python dependencies required in this chapter, as well as Chapter 22.
Code
req <- c('access==1.1.9', 'affine==2.4.0', 'anndata==0.9.2', 'anndata2ri==1.3.1', 'attrs==23.1.0', 'backports.zoneinfo==0.2.1', 'beautifulsoup4==4.12.2', 'certifi==2023.11.17', 'cffi==1.16.0', 'charset-normalizer==3.3.2', 'click==8.1.7', 'click-plugins==1.1.1', 'cligj==0.7.2', 'commot==0.0.3', 'contourpy==1.1.1', 'cycler==0.12.1', 'decorator==4.4.2', 'deprecation==2.1.0', 'esda==2.4.3', 'fiona==1.9.5', 'fonttools==4.45.1', 'gensim==4.3.2', 'geopandas==0.13.2', 'get-annotations==0.1.2', 'giddy==2.3.4', 'h5py==3.10.0', 'idna==3.4', 'igraph==0.10.8', 'importlib-metadata==6.8.0', 'importlib-resources==6.1.1', 'inequality==1.0.0', 'Jinja2==3.1.2', 'joblib==1.3.2', 'karateclub==1.3.3', 'kiwisolver==1.4.5', 'leidenalg==0.10.1', 'Levenshtein==0.23.0', 'libpysal==4.7.0', 'llvmlite==0.41.1', 'mapclassify==2.6.0', 'MarkupSafe==2.1.3', 'matplotlib==3.7.4', 'mgwr==2.1.2', 'momepy==0.6.0', 'mpmath==1.3.0', 'natsort==8.4.0', 'networkx==2.6.3', 'numba==0.58.1', 'numpy==1.22.4', 'packaging==23.2', 'pandas==1.3.5', 'patsy==0.5.3', 'Pillow==10.1.0', 'platformdirs==4.0.0', 'plotly==5.18.0', 'pointpats==2.4.0', 'POT==0.9.1', 'PuLP==2.7.0', 'pycparser==2.21', 'pygeos==0.14', 'PyGSP==0.5.1', 'pynndescent==0.5.11', 'pyparsing==3.1.1', 'pyproj==3.5.0', 'pysal==23.7', 'python-dateutil==2.8.2', 'python-igraph==0.10.8', 'python-Levenshtein==0.23.0', 'python-louvain==0.16', 'pytz==2023.3.post1', 'quantecon==0.7.1', 'rapidfuzz==3.5.2', 'rasterio==1.3.9', 'rasterstats==0.19.0', 'requests==2.31.0', 'rpy2==3.5.14', 'Rtree==1.1.0', 'scanpy==1.9.6', 'scikit-learn==1.3.2', 'scipy==1.10.1', 'seaborn==0.12.2', 'segregation==2.5', 'session-info==1.0.0', 'setuptools==68.2.2', 'shapely==2.0.2', 'simplejson==3.19.2', 'six==1.16.0', 'smart-open==6.4.0', 'snuggs==1.4.7', 'soupsieve==2.5', 'spaghetti==1.7.4', 'spglm==1.0.8', 'spint==1.0.7', 'splot==1.1.5.post1', 'spopt==0.5.0', 'spreg==1.4', 'spvcm==0.3.0', 'statsmodels==0.14.0', 'stdlib-list==0.10.0', 'sympy==1.12', 'tenacity==8.2.3', 'texttable==1.7.0', 'threadpoolctl==3.2.0', 'tobler==0.9.0', 'tqdm==4.66.1', 'tzlocal==5.2', 'umap-learn==0.5.5', 'urllib3==2.1.0', 'zipp==3.17.0')
install_python(version=ver <- "3.8.18")
virtualenv_create(envname=env <- "OSTA", python=ver, packages=req)
use_virtualenv(env, required=TRUE)7.3.2 Using Conda
Alternatively, an existing Conda environment – including the corresponding Python installation and software dependencies – could be used as follows:
Code
# path to Conda binary
bin <- ".../bin/conda"
options(reticulate.conda_binary=bin)
# activate environment
use_condaenv("<name>")
7.3.3 Using basilisk
Alternatively, we can use basilisk to install and manage Python environments. This is an alternative to setting up a Conda environment with an existing Python installation. Using basilisk provides a self-contained environment, which helps avoid problems due to interactions with other environments on your system, and provides a higher level of reproducibility. This is especially useful for package development by advanced users. However, setup may be more challenging in some cases. A minimal example is provided here for completeness:
Code
# set up environment using 'basilisk'
env <- BasiliskEnvironment(
pkgname="base",
envname="basilisk",
pip="numpy==2.4.0",
packages="python=3.12")
# activate virtual environment
use_virtualenv(obtainEnvironmentPath(env))7.4 Interfacing between languages
7.4.1 SingleCellExperiment
7.4.1.1 Calling Python
After configuring Python, R commands can now be run using reticulate; for more details on syntax, see here.
Code
import scanpy
id = "V1_Mouse_Brain_Sagittal_Posterior"
ad = scanpy.datasets.visium_sge(sample_id=id)##
0%| | 0.00/9.26M [00:00<?, ?B/s]
75%|#######5 | 6.98M/9.26M [00:00<00:00, 73.1MB/s]
100%|##########| 9.26M/9.26M [00:00<00:00, 84.9MB/s]
##
0%| | 0.00/20.1M [00:00<?, ?B/s]
33%|###3 | 6.69M/20.1M [00:00<00:00, 69.8MB/s]
100%|##########| 20.1M/20.1M [00:00<00:00, 132MB/s]Code
ad.var_names_make_unique()
ad## AnnData object with n_obs × n_vars = 3355 × 32285
## obs: 'in_tissue', 'array_row', 'array_col'
## var: 'gene_ids', 'feature_types', 'genome'
## uns: 'spatial'
## obsm: 'spatial'7.4.1.2 Continuing in R
We can access any of the variables above in R. For basic outputs, this works out of the box:
Code
range(py$ad$obs$array_row)## [1] 6 72reticulate also supports a few direct type conversions (e.g. dictionary \(\leftrightarrow\) named list). In the example demonstrated here, we use anndataR to convert from AnnData to SingleCellExperiment:
Code
(sce <- py$ad$as_SingleCellExperiment(x_mapping="counts"))## class: SingleCellExperiment
## dim: 32285 3355
## metadata(1): spatial
## assays(1): counts
## rownames(32285): Xkr4 Gm1992 ... AC234645.1 AC149090.1
## rowData names(3): gene_ids feature_types genome
## colnames(3355): AAACAAGTATCTCCCA-1 AAACACCAATAACTGC-1 ...
## TTGTTTCATTAGTCTA-1 TTGTTTCCATACAACT-1
## colData names(3): in_tissue array_row array_col
## reducedDimNames(1): spatial
## mainExpName: NULL
## altExpNames(0):7.4.1.3 Back to Python
We can also do the reverse, i.e., go from R’s SingleCellExperiment to Python’s AnnData:
Code
(ad <- as_AnnData(sce, x_mapping="counts"))## InMemoryAnnData object with n_obs × n_vars = 3355 × 32285
## obs: 'in_tissue', 'array_row', 'array_col'
## var: 'gene_ids', 'feature_types', 'genome'
## uns: 'spatial'
## obsm: 'spatial'7.4.2 SpatialExperiment
Since the SpatialExperiment class extends SingleCellExperiment (see Chapter 3), conversion operations discussed above are also applicable to SpatialExperiment. However, to accomplish a full conversion from the AnnData object, we need to manually insert the spatial information using reticulate directly.
7.4.2.1 Starting in R
For this use case with SpatialExperiment, we will use the dataset from Janesick et al. (2023), which includes Visium measurements on human breast cancer tissue.
Code
id <- "Visium_HumanBreast_Janesick"
pa <- OSTA.data_load(id)
dir.create(td <- tempfile())
unzip(pa, exdir = td)
obj <- TENxVisium(
spacerangerOut=file.path(td, "outs"),
format="h5",
images="lowres")
(spe <- VisiumIO::import(obj))We also need to parse the original scaling information (i.e., scale factor) for spots and images available in the standard Visium output. We will use this later during conversion.
We again use the as_AnnData function from anndataR from the previous example, and convert the SingleCellExperiment-relevant components of the SpatialExperiment object to an AnnData object.
Code
mtx <- as(assay(spe), "dgCMatrix")
assay(spe) <- mtx
(ad <- as_AnnData(spe))## InMemoryAnnData object with n_obs × n_vars = 4992 × 18085
## obs: 'in_tissue', 'array_row', 'array_col', 'sample_id'
## var: 'ID', 'Symbol', 'Type'
## uns: 'resources', 'spatialList'
## layers: 'counts'We can now populate the uns and obsm components of the AnnData object with spatial coordinates and images. We start with the coordinates.
Code
spatialCoordsNames(spe) <- c("x", "y")
obsm <- list(spatial=spatialCoords(spe))Now, let’s create the uns component. The list of uns should be composed of as many samples as the images in the SpatialExperiment object. Also, each sample entry in the list should have two elements, one for the image and the other for the scaling information.
Code
# get image metadata
imgdata <- imgData(spe)
# get image
img <- imgRaster(spe)
img <- apply(img, c(1, 2), \(x) col2rgb(x))
img <- aperm(img, perm=c(2, 3, 1))
img <- img / 255
# create uns
uns <- list(images=list(lowres=img), scalefactors=sfs)
uns <- list(spatial=setNames(list(uns), imgdata$sample_id))Now let’s insert the components to the AnnData object, and write back to an .h5ad file.
Code
ad$obs$library_id <- imgdata$sample_id
ad$obsm <- obsm
ad$uns <- uns
ad$write_h5ad("spe.h5ad")7.4.2.2 Calling Python
Now that we have converted the SpatialExperiment object to AnnData format, we can run Python code per usual (e.g., continue analysis using squidpy). As a proof of concept, we here visualize an exemplary gene’s counts spatially:
Code
import anndata
import matplotlib
import matplotlib.pyplot as plt
ad = anndata.read_h5ad("spe.h5ad")
ad.var_names = ad.var["Symbol"].astype(str)
xy = ad.obsm["spatial"]
z = ad[:,"ERBB2"].layers["counts"].toarray()
plt.scatter(xy[:,0], xy[:,1], c=z, s=5, cmap="turbo")
plt.gca().set_aspect("equal")
plt.title("ERBB2")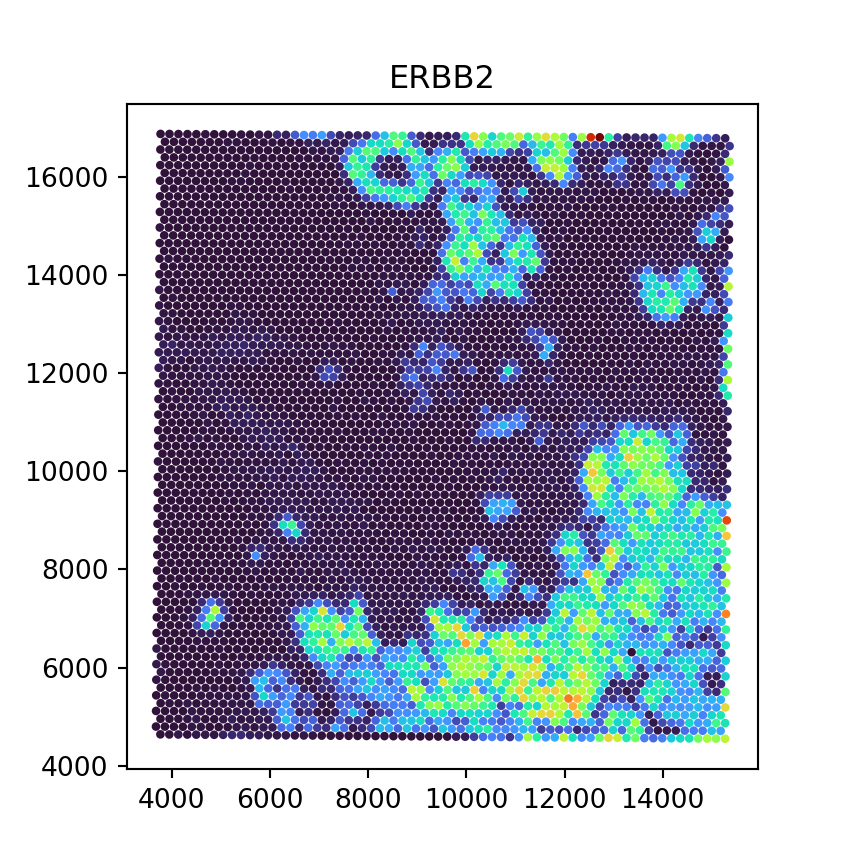
7.5 Appendix
Following are links to several key packages and tools relating to R-Python interoperability, which were mentioned in the sections above:
anndataR (Deconinck et al. 2025): R package and community project to work with
AnnDataobjects in R, including conversion to and fromSingleCell/SpatialExperimentandSeuratobjectszellkonverter (Zappia et al. 2020): R package to convert between
AnnDataandSingleCellExperimentobjects, as well as reading from and writing to H5ADreticulate (Ushey, Allaire, and Tang 2017): R package and framework to call Python and run Python code in R, translate between R and Python objects, and manage virtual and Conda environments
basilisk (Lun 2022): R package to install and manage Python environments in R packages and sessions