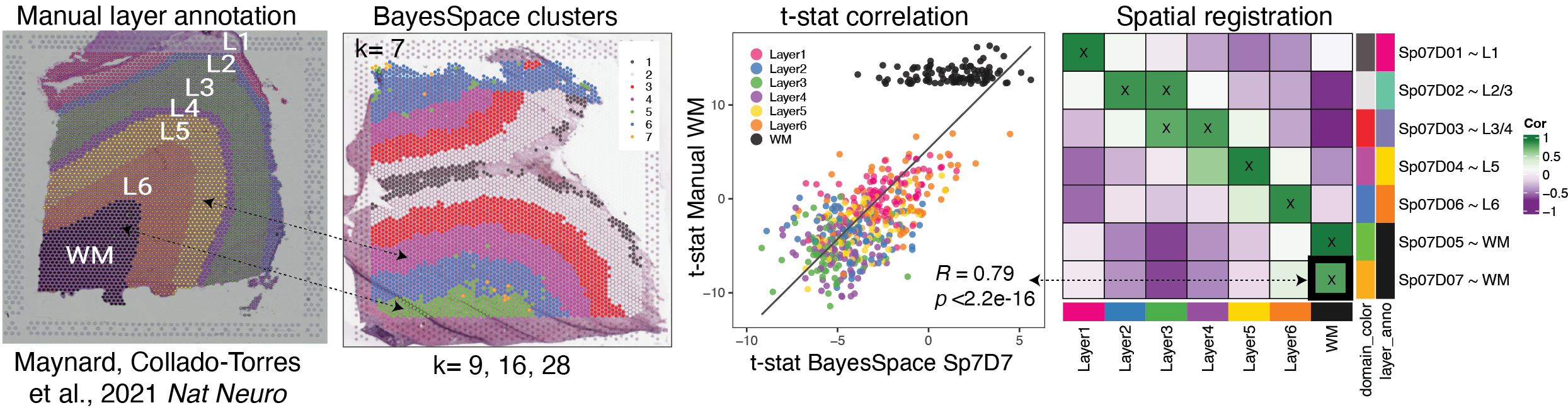
{width=100%}
# How to run Spatial Registration with `spatialLIBD` tools ## Introduction. In this example we will utilize the human DLPFC 10x Genomics Visium dataset from Maynard, Collado-Torres et al. `r Citep(bib[['spatialLIBDpaper']])` as the **reference**. This data contains manually annotated features: the **six cortical layers + white matter** present in the DLPFC. We will use the pre-calculated enrichment statistics for the layers, which are available from `r Biocpkg("spatialLIBD")`.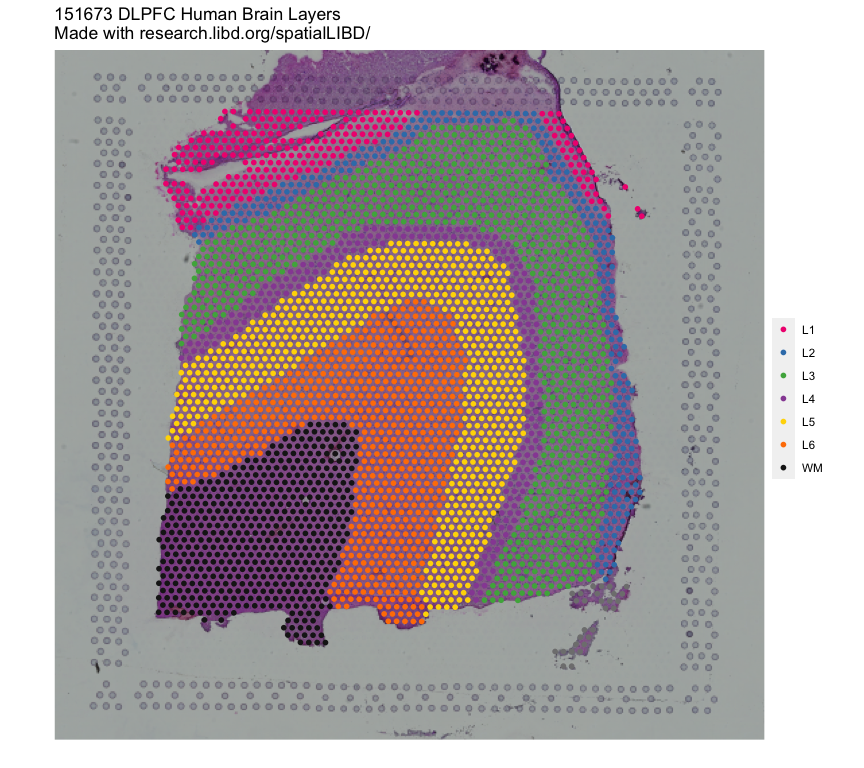{width=100%}
The **query** dataset will be the DLPFC single nucleus RNA-seq (snRNA-seq) data from `r Citep(bib[['tran2021']])`. We will compare the gene expression in the cell type populations of the **query** dataset to the annotated **layers** in the **reference**. ## Important Notes ### Required knowledge It may be helpful to review _Introduction to spatialLIBD_ vignette available through [GitHub](http://research.libd.org/spatialLIBD/articles/spatialLIBD.html) or [Bioconductor](https://bioconductor.org/packages/spatialLIBD) for more information about this data set and R package. ### Citing `spatialLIBD` We hope that `r Biocpkg("spatialLIBD")` will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you! ```{r "citation"} ## Citation info citation("spatialLIBD") ``` ## Setup ### Install `spatialLIBD` ```{r "install", eval = FALSE} if (!requireNamespace("BiocManager", quietly = TRUE)) { install.packages("BiocManager") } BiocManager::install("spatialLIBD") ## Check that you have a valid Bioconductor installation BiocManager::valid() ``` ### Load required packages ```{r "start", message=FALSE} library("spatialLIBD") library("SingleCellExperiment") ``` ## Download Data ### Spatial Reference The reference data is easily accessed through `r Biocpkg("spatialLIBD")`. The modeling results for the annotated layers is already calculated and can be accessed with the `fetch_data()` function. This data contains the results form three models (anova, enrichment, and pairwise), we will use the **enrichment** results for spatial registration. The tables contain the $t$-statistics, p-values, and gene ensembl ID and symbol. ```{r "fetch_refrence"} ## get reference layer enrichment statistics layer_modeling_results <- fetch_data(type = "modeling_results") layer_modeling_results$enrichment[1:5, 1:5] ``` ### Query Data: snRNA-seq For the query data set, we will use the public single nucleus RNA-seq (snRNA-seq) data from `r Citep(bib[['tran2021']])` can be accessed on [github](https://github.com/LieberInstitute/10xPilot_snRNAseq-human#processed-data). This data is also from postmortem human brain DLPFC, and contains gene expression data for 11k nuclei and 19 cell types. We will use `BiocFileCache()` to cache this data. It is stored as a `SingleCellExperiment` object named `sce.dlpfc.tran`, and takes 1.01 GB of RAM memory to load. ```{r "download_sce_data"} # Download and save a local cache of the data available at: # https://github.com/LieberInstitute/10xPilot_snRNAseq-human#processed-data bfc <- BiocFileCache::BiocFileCache() url <- paste0( "https://libd-snrnaseq-pilot.s3.us-east-2.amazonaws.com/", "SCE_DLPFC-n3_tran-etal.rda" ) local_data <- BiocFileCache::bfcrpath(url, x = bfc) load(local_data, verbose = TRUE) ``` DLPFC tissue consists of many cell types, some are quite rare and will not have enough data to complete the analysis ```{r "check_cell_types"} table(sce.dlpfc.tran$cellType) ``` The data will be pseudo-bulked over `donor` x `cellType`, it is recommended to drop groups with < 10 nuclei (this is done automatically in the pseudobulk step). ```{r "donor_x_cellType"} table(sce.dlpfc.tran$donor, sce.dlpfc.tran$cellType) ``` ## Get Enrichment statistics for snRNA-seq data `spatialLIBD` contains many functions to compute `modeling_results` for the query sc/snRNA-seq or spatial data. **The process includes the following steps** 1. `registration_pseudobulk()`: Pseudo-bulks data, filter low expressed genes, and normalize counts 2. `registration_mod()`: Defines the statistical model that will be used for computing the block correlation 3. `registration_block_cor()` : Computes the block correlation using the sample ID as the blocking factor, used as correlation in eBayes call 2. `registration_stats_enrichment()` : Computes the gene enrichment $t$-statistics (one group vs. All other groups) The function `registration_wrapper()` makes life easy by wrapping these functions together in to one step! ```{r "run_registration_wrapper"} ## Perform the spatial registration sce_modeling_results <- registration_wrapper( sce = sce.dlpfc.tran, var_registration = "cellType", var_sample_id = "donor", gene_ensembl = "gene_id", gene_name = "gene_name" ) ``` ## Extract Enrichment t-statistics ```{r "extract_t_stats"} ## extract t-statics and rename registration_t_stats <- sce_modeling_results$enrichment[, grep("^t_stat", colnames(sce_modeling_results$enrichment))] colnames(registration_t_stats) <- gsub("^t_stat_", "", colnames(registration_t_stats)) ## cell types x gene dim(registration_t_stats) ## check out table registration_t_stats[1:5, 1:5] ``` ## Correlate statsics with Layer Reference ```{r "layer_stat_cor"} cor_layer <- layer_stat_cor( stats = registration_t_stats, modeling_results = layer_modeling_results, model_type = "enrichment", top_n = 100 ) cor_layer ``` # Explore Results Now we can use these correlation values to learn about the cell types. ## Create Heatmap of Correlations We can see from this heatmap what layers the different cell types are associated with. * Oligo with WM * Astro with Layer 1 * Excitatory neurons to different layers of the cortex * Weak associate with Inhibitory Neurons ```{r layer_cor_plot} layer_stat_cor_plot(cor_layer, max = max(cor_layer)) ``` ## Annotate Cell Types by Top Correlation We can use `annotate_registered_clusters` to create annotation labels for the cell types based on the correlation values. ```{r "annotate"} anno <- annotate_registered_clusters( cor_stats_layer = cor_layer, confidence_threshold = 0.25, cutoff_merge_ratio = 0.25 ) anno ``` # Reproducibility The `r Biocpkg("spatialLIBD")` package `r Citep(bib[["spatialLIBD"]])` was made possible thanks to: * R `r Citep(bib[["R"]])` * `r Biocpkg("BiocStyle")` `r Citep(bib[["BiocStyle"]])` * `r CRANpkg("knitr")` `r Citep(bib[["knitr"]])` * `r CRANpkg("RefManageR")` `r Citep(bib[["RefManageR"]])` * `r CRANpkg("rmarkdown")` `r Citep(bib[["rmarkdown"]])` * `r CRANpkg("sessioninfo")` `r Citep(bib[["sessioninfo"]])` * `r CRANpkg("testthat")` `r Citep(bib[["testthat"]])` This package was developed using `r BiocStyle::Biocpkg("biocthis")`. Code for creating the vignette ```{r createVignette, eval=FALSE} ## Create the vignette library("rmarkdown") system.time(render("guide_to_spatial_registration.Rmd", "BiocStyle::html_document")) ## Extract the R code library("knitr") knit("guide_to_spatial_registration.Rmd", tangle = TRUE) ``` Date the vignette was generated. ```{r reproduce1, echo=FALSE} ## Date the vignette was generated Sys.time() ``` Wallclock time spent generating the vignette. ```{r reproduce2, echo=FALSE} ## Processing time in seconds totalTime <- diff(c(startTime, Sys.time())) round(totalTime, digits = 3) ``` `R` session information. ```{r reproduce3, echo=FALSE} ## Session info library("sessioninfo") options(width = 120) session_info() ``` # Bibliography This vignette was generated using `r Biocpkg("BiocStyle")` `r Citep(bib[["BiocStyle"]])` with `r CRANpkg("knitr")` `r Citep(bib[["knitr"]])` and `r CRANpkg("rmarkdown")` `r Citep(bib[["rmarkdown"]])` running behind the scenes. Citations made with `r CRANpkg("RefManageR")` `r Citep(bib[["RefManageR"]])`. ```{r vignetteBiblio, results = "asis", echo = FALSE, warning = FALSE, message = FALSE} ## Print bibliography PrintBibliography(bib, .opts = list(hyperlink = "to.doc", style = "html")) ```